# 植物空间转录组分析 1:Seurat 基本流程
# 植物空间转录组分析 2:STEEL+Seurat
# 前言
时隔半年的更新,这次的主题是单细胞 hdWGCNA 分析,文章刚刚被接收,目前在植物空间转录组已经有文章使用了该方法,基本的内容就是分 module 然后去看每个 module 的 hub 基因,或者做做 GO 富集之类的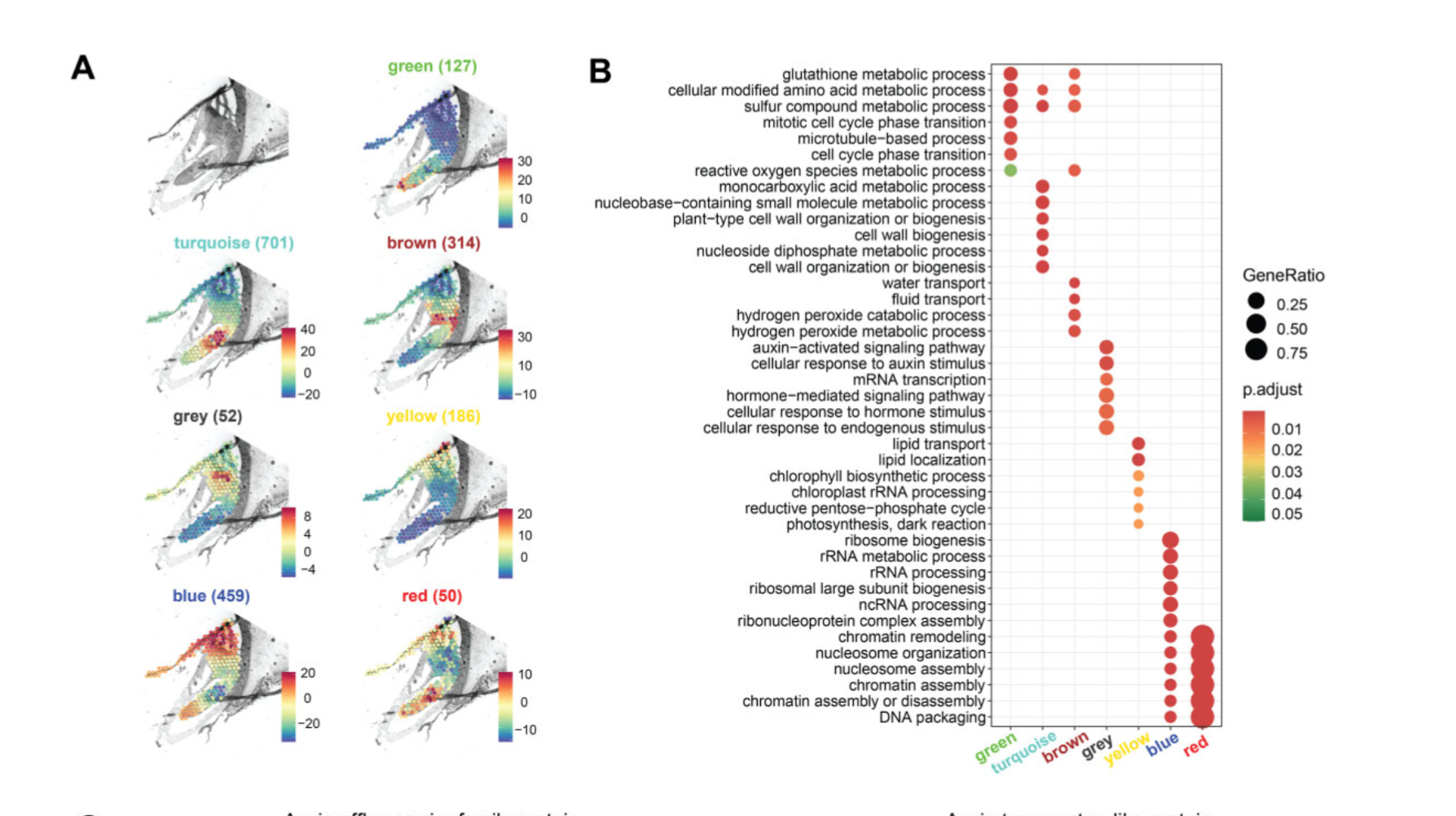
# R 包安装以及数据准备
本次分析还是以兰花的空间转录组为基础,具体的数据参考以前的推文
# install BiocManager | |
install.packages("BiocManager") | |
# install Bioconductor core packages | |
BiocManager::install() | |
# install additional packages: | |
install.packages(c("Seurat", "WGCNA", "igraph", "devtools")) | |
devtools::install_github('smorabit/hdWGCNA', ref='dev') |
大家可能会因为网络问题无法下载,我直接将压缩包发给大家
## 数据准备 | |
## 将 spot 坐标信息加到 metadata 中 | |
# make a dataframe containing the image coordinates for each sample | |
image_df <- do.call (rbind, lapply (names (orc1_new@images), function (x){ | |
orc1_new@images [[x]]@coordinates | |
})) | |
# merge the image_df with the Seurat metadata | |
new_meta <- merge (orc1_new@meta.data, image_df, by='row.names') | |
# fix the row ordering to match the original seurat object | |
rownames (new_meta) <- new_meta$Row.names | |
ix <- match (as.character (colnames (orc1_new)), as.character (rownames (new_meta))) | |
new_meta <- new_meta [ix,] | |
# add the new metadata to the seurat object | |
orc1_new@meta.data <- new_meta |
# 构建 metaspots
## Here we set up the data for hdWGCNA and run MetaspotsByGroups. | |
subdata <- subset(orc1_new, idents = c(19,21,37,38,39,40)) | |
subdata <- SCTransform(subdata, assay = "Spatial", return.only.var.genes = FALSE, verbose = FALSE) | |
subdata <- RunPCA(subdata, features = VariableFeatures(subdata)) | |
subdata <- SetupForWGCNA( | |
subdata, | |
features = VariableFeatures(subdata), | |
wgcna_name = "SCT" | |
) | |
subdata <- MetacellsByGroups( | |
seurat_obj = subdata, | |
group.by = c("seurat_clusters"), | |
k = 5, | |
max_shared= 10, | |
min_cells = 6, | |
reduction = 'pca', | |
ident.group = 'seurat_clusters', | |
slot = 'scale.data', | |
assay = 'SCT' | |
) | |
m_obj <- GetMetacellObject(subdata) | |
m_obj | |
An object of class Seurat | |
14006 features across 180 samples within 1 assay | |
Active assay: SCT (14006 features, 0 variable features) |
这里需要注意,一般空间分析推荐使用的是 MetaspotsByGroups 函数,我尝试了一遍 softpower 只有 1,module 只有两个,所以又使用了 MetacellsByGroups 函数尝试。
# 共表达网络分析
# set up the expression matrix, set group.by and group_name to NULL to include all spots | |
subdata <- SetDatExpr( | |
subdata, | |
group.by=NULL, | |
group_name = NULL | |
) | |
# test different soft power thresholds | |
subdata <- TestSoftPowers(subdata) | |
plot_list <- PlotSoftPowers(subdata) | |
wrap_plots(plot_list, ncol=2) |
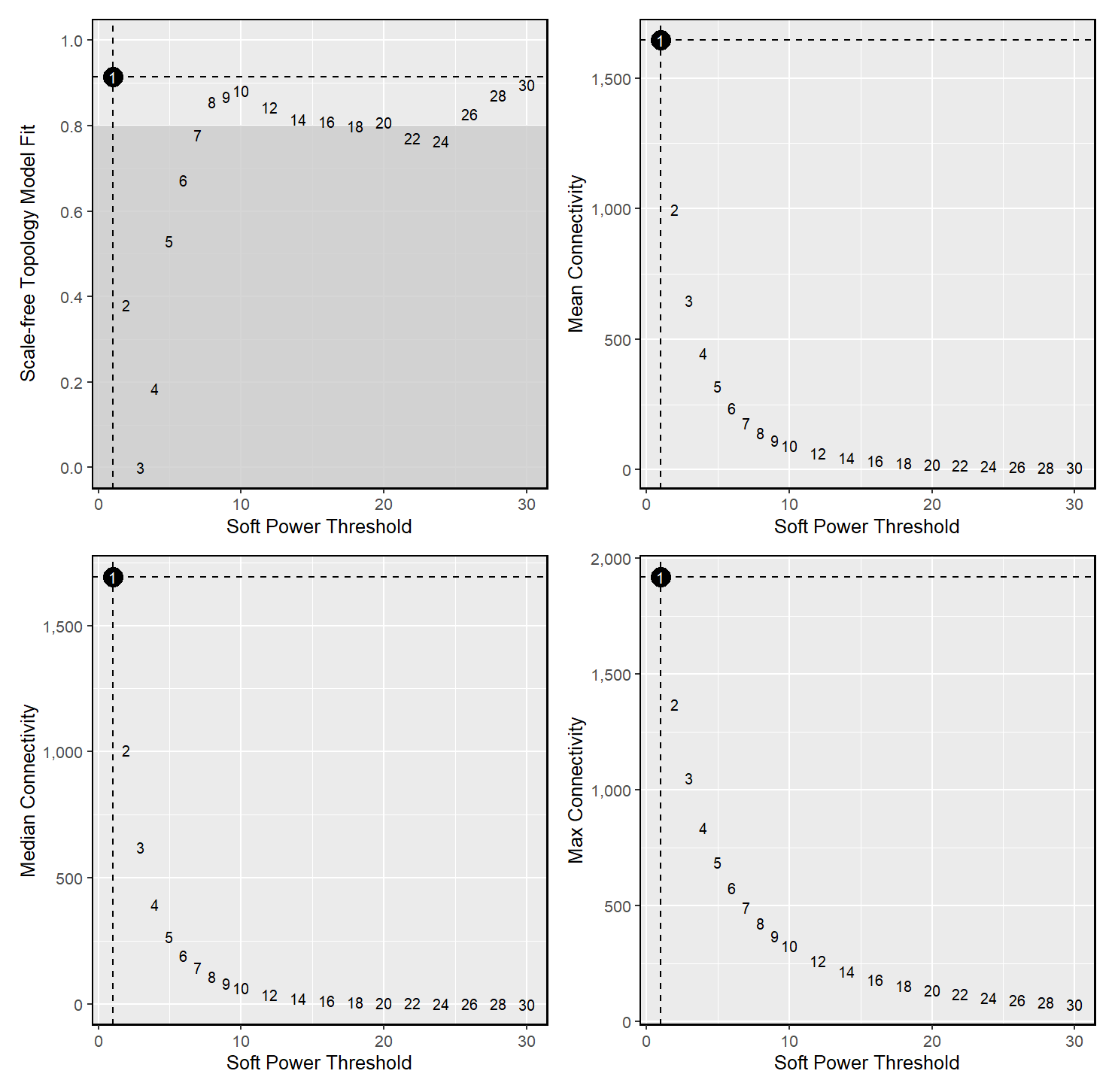
# construct co-expression network:
subdata <- ConstructNetwork(
subdata,
soft_power = 1,
tom_name='test',
overwrite_tom=TRUE
)
# plot the dendrogram
PlotDendrogram(subdata, main='Spatial hdWGCNA dendrogram')
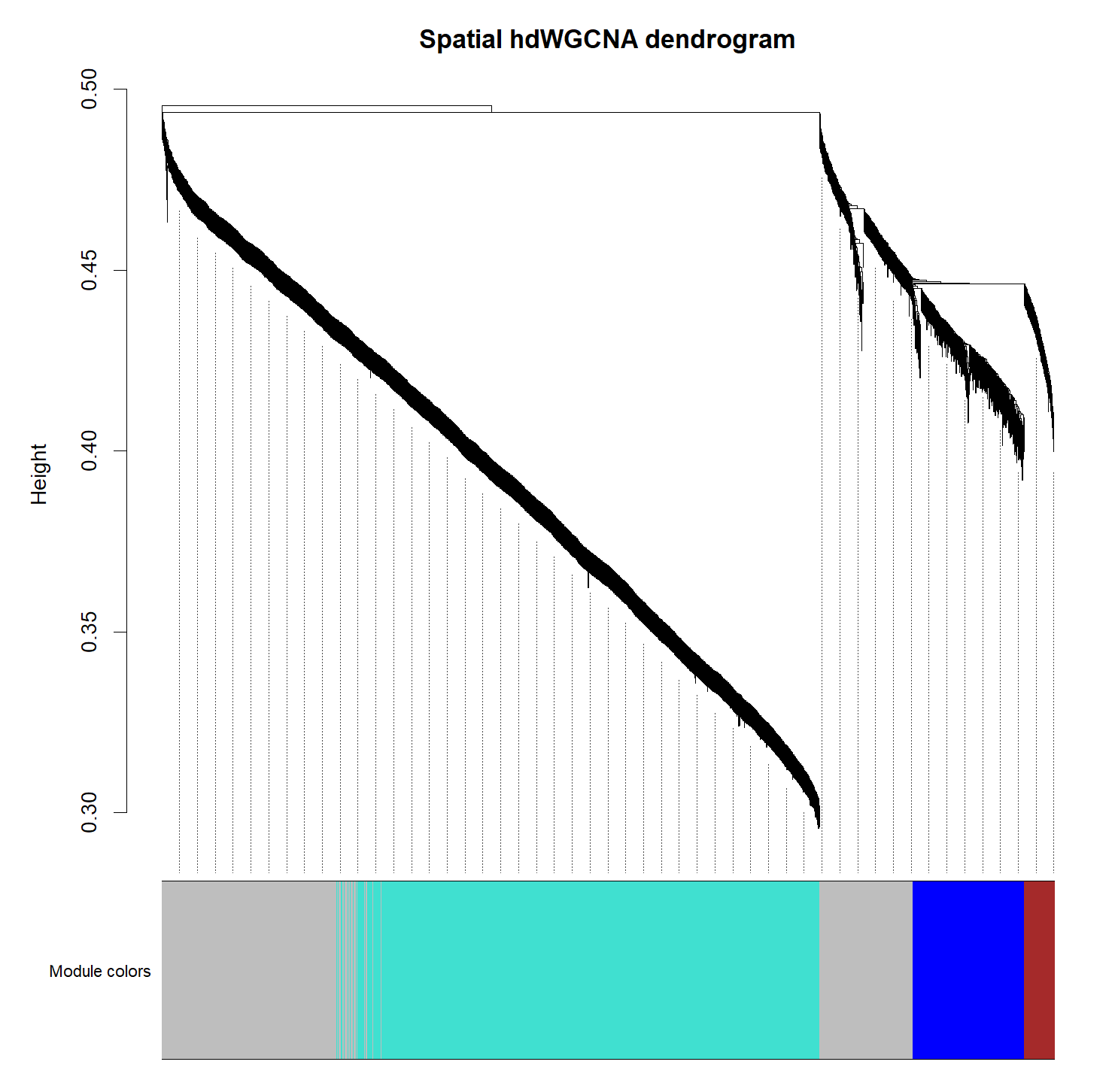
# compute module eigengenes
subdata <- ModuleEigengenes(subdata) | |
subdata <- ModuleConnectivity(subdata) | |
modules <- GetModules(subdata) %>% subset(module != 'grey') | |
head(modules[,1:3]) | |
gene_name module color | |
PAXXG297280 PAXXG297280 turquoise turquoise | |
PAXXG074500 PAXXG074500 blue blue | |
PAXXG239080 PAXXG239080 turquoise turquoise | |
PAXXG067750 PAXXG067750 turquoise turquoise | |
PAXXG327430 PAXXG327430 turquoise turquoise | |
PAXXG311150 PAXXG311150 blue blue |
模块数目比较少也是正常的
# Data visualization
tom<-GetTOM(subdata) | |
##Data visualization | |
##Here we visualize module eigengenes using the Seurat functions DotPlot and SpatialFeaturePlot. | |
##For network visualization, please refer to the Network visualization tutorial. | |
# get module eigengenes and gene-module assignment tables | |
MEs <- GetMEs(subdata) | |
modules <- GetModules(subdata) | |
mods <- levels(modules$module) | |
write.table(file="modules.xls",modules,row.names = F,sep="\t") | |
# plot with Seurat's DotPlot function | |
p <- DotPlot(subdata, features=mods, group.by = 'seurat_clusters', dot.min=0.1) | |
# flip the x/y axes, rotate the axis labels, and change color scheme: | |
p <- p + | |
coord_flip() + | |
RotatedAxis() + | |
scale_color_gradient2(high='red', mid='grey95', low='blue') + | |
xlab('') + ylab('') | |
p | |
p <- SpatialFeaturePlot( | |
subdata, | |
features = mods, | |
ncol = 4, | |
pt.size.factor = 3.5 | |
) | |
p |


{.gallery data-height="300"}
分别用两种方法展示
# module network plots
分为两种,一种每个模块的 hubgene,一种是全部模块的 hubgene
## Individual module network plots | |
ModuleNetworkPlot(subdata, | |
plot_size = c(10, 10), | |
vertex.label.cex = 0.6 | |
) | |
hub_df <- GetHubGenes(subdata, n_hubs = 25) | |
write.table(file="hub_df.xls",hub_df,row.names = F,sep="\t") | |
## hubgene network | |
HubGeneNetworkPlot( | |
subdata, | |
n_hubs = 4, n_other=6, | |
edge_prop = 0.75, | |
mods = 'all', | |
hub.vertex.size = 4, | |
other.vertex.size = 1, | |
edge.alpha = 0.5 | |
) |


# 总结
目前只是初步上手,其实这个方法主要的目的还是根据表达对基因进行聚类,再加个功能富集可能会更好,后续还有 UMAP to co-expression networks 流程,但因为我在做的时候效果比较差所以没放。